Just as species evolve through mutation and selection acting on individuals in a population, cancer cells and tumors evolve by mutation and selection acting on cells within the tumor. Normal cells divide and mutate over time into subpopulations of cancerous cells – or subclones – with distinct mutation patterns. These expand and come together to form a tumor with many heterogenous subclones that can drive a cancer’s rate of growth and determine its resistance to therapy.
That’s the foundation of cancer phylogenetics, a relatively new research field that seeks to understand the evolution of tumors over time so they can be more effectively treated.
Researchers create evolutionary “trees” to understand tumor evolution, but as with many new methods of scientific discovery, the tools used to build histories of tumor evolution can be difficult to use, and there is no standard framework for analyzing and visualizing the data collected through phylogeny.

There are different algorithms for different assay types, and matching methods of analysis, specifically to the types of data collected, can be difficult. There’s a need for best practices to analyze and build histories of tumor evolution and then share them to build a consensus framework, where results can be available to an entire community of researchers.
Charles Blatti, PhD, research scientist, NCSA Visual Analytics Group
Blatti is a co-principal investigator for a project that’s building that framework, pulling together open source tools to analyze and annotate tumor data and then merging those results into a visualization tool that allows clinical researchers to view results easily. The visualizations enable researchers to easily see the history and distribution of mutations, dive deeper into information about gene variants, make predictions about tumor growth, and target therapies to best suit a particular patient’s condition.
“Phylogeny requires measuring the frequency of mutations in different cells,” said Nicholas Chia, PhD, an associate professor at Mayo Clinic who studies colorectal cancer. “This can require high coverage sequencing or single-cell sequencing to accurately assess. Both have limitations.”
Chia is a partner in the NCSA project, providing patient data and feedback on the new tools, as is Zeynep Madak-Erdogan, PhD, an associate professor of food science and nutrition at the University of Illinois Urbana-Champaign, who studies breast cancer. The project received seed funding from the Cancer Center at Illinois (CCIL) in 2021 and won another round of funding from CCIL in 2022. Mohammed El-Kebir, PhD, assistant professor in the UIUC computer science department, served as PI on the seed grant and he and his graduate students lead the efforts to incorporate phylogenetic algorithms and methods into the analysis workflow.
“Workflows are extremely important in bioinformatics,” said Jessica Saw, MD, PhD, a research scientist in NCSA’s Visual Analytics Group. “Usually, the researcher must download a bunch of different applications for different functions and an output from one application will become an input for another. Workflows combine them all, from making sure the data is clean to analysis and visualization.”
The goal, she said, is to make executing a workflow simple enough that doctors and clinical researchers can focus on their work and exploring their data rather than writing code and learning to execute different software applications.
Tools for Analysis and Real-Time Interaction
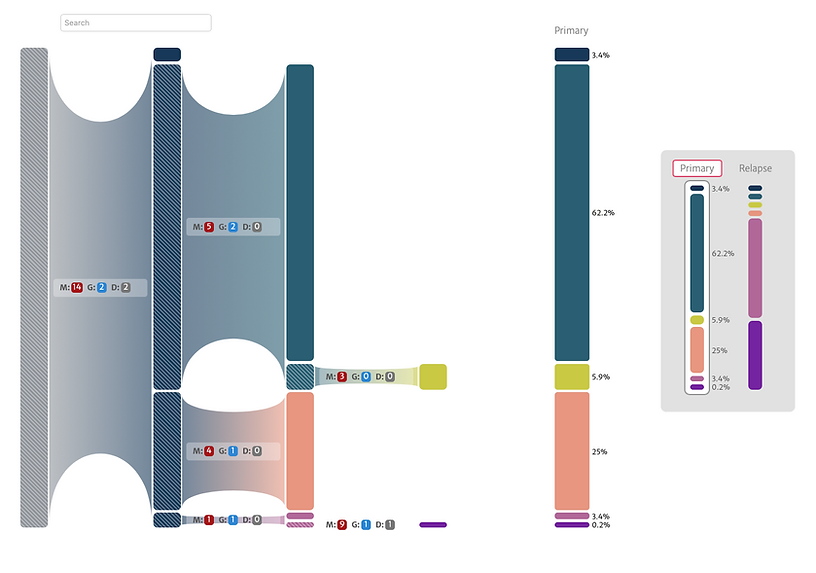
The NCSA project has two components: PhyloFlow combines proven open source applications into one easy-to-use tool that provides end-to-end cancer phylogenetic analysis; PhyloDiver takes the data from PhyloFlow and develops visualizations that the end user can interact with in real-time.
The tools incorporated into PhyloFlow include the Variant Effect Predictor (VEP), an open-source tool for annotating and filtering genomic variants; PyClone, a statistical model used to infer the structure of clonal cell populations in tumors; SPRUCE, an algorithm that can describe the evolution of mutations in a tumor when given sequencing data; and JSON, an open-file format for data interchange that uses text to store and transmit data objects.
“We’re aiming for maturity in this type of analysis, so there are standard steps” said Blatti. “With a common workflow users can know their data has been treated the same way, and making comparisons becomes easier.”
After the phylogenetic data is processed through PhyloFlow, it creates an evolutionary tree, or phylogeny, to better understand the heterogenous nature of tumors. The PhyloDiver visualization tool helps users see these results in a more intuitive way and with greater detail.

Instead of viewing data as a spreadsheet, users see color-coded bars, with the occurrence of mutated variants in different tumor subclones marked. With a simple mouse click, users can dig deeper and see more information about variants, including the distribution of the mutation and drugs with known sensitivities to it.
Jessica Saw, research scientist in the Visual Analytics Group
“Generally, doctors use two-dimensional fish plots to see this kind of information,” said Lisa Gatzke, a senior UX research designer who works on developing the PhyloFlow interface. “We wanted our tool to be more interactive so you can get more information on the mutation.”
Some of that interactivity includes the ability to overlay other information about the mutation, being able to add annotations or link to existing annotations, finding information about single nucleotide variants (SNVs) and their severity, comparing output from different phylogenetic trees, and discovering information about drugs that can treat the tumor. The tools help researchers track changes in mutations over time and make predictions about expected clonal growth – something that could help them understand why some cancers stay in remission and some come back.
“Our goal is to know what information clinicians most need to see to guide their decision making,” said Gatzke. “We want to give them the answers they need in the most efficient way possible.”
The NCSA visualizations not only put data into an intuitive format, they also provide much greater detail, according to Saw. For example, after a breast cancer diagnosis, a pathologist will look at a sample of the tumor cells under a microscope to see their attributes and an oncologist will use these results to determine the best treatment options. Generally, the tumor types are organized into a few broad buckets.
“It’s like looking at a picture very closely and being able to see the individual pixels,” said Saw.
“At first glance, it may seem that all tumors of a given subtype are created equal but within each individual tumor, there are many, many subtypes with different evolutionary backgrounds. If we can see more of those differences and understand how they affect a patient, we can use this information to predict how a patient responds to treatment and long-term prognosis.”
Enabling researchers and doctors to see how cancerous mutations change over time can also provide clues about whether tumors are likely to mutate further, according to Blatti.
There are the initial mutations that you can hopefully treat with drugs, but what we often see is that over time, another mutation takes over, and you need to find a way to treat that. We hope we can help doctors anticipate what the path of the cancer might be by making it easier to track its evolution.
PhyloFlow and PhyloDiver are being tested now by Chia and Madak-Erdogan. The team continues to refine the tools and hopes to secure more funding that would enable them to share the tools with more medical professionals to get more feedback. Other key contributors to the project are Peter Groves, NCSA senior software engineer, and Chuanyi Zhang, a computer science graduate student in El-Kebir’s lab, who carry out the PhyloFlow workflow development; as well as NCSA research software engineers Chad Olson and Matt Berry who implement the PhyloDiver application design and development.
If widely adopted, PhyloFlow and PhyloDiver could help researchers understand and track tumor evolution and enable doctors to develop more personalized cancer treatment plans.
“The main benefit is that it makes tumor phylogeny accessible to a broader population of researchers that would normally not have the expertise or computational infrastructure to carry out these important studies,” said Chia. “We hope that by making these tools more accessible we will enable a new generation of research that includes cancer evolution as part of its general toolbox.”