
Have you ever taken a prescribed medicine to resolve a health issue, only for the treatment to fail? Perhaps you’re among the unlucky low percentage of people on weight loss drugs who can’t seem to lose a single pound. The lack of efficacy in your treatments may be due to your unique genetic profile. Our specific genes can have many subtle effects on our health that don’t necessarily fit the average. Two researchers from Professor Hongyu Zhao’s lab at Yale University are working on AI tools to change that, and they’ve used NCSA’s Delta supercomputer to support their projects.
Decoding Your Unique Blueprint

Tianyu Liu is a Ph.D. candidate at Yale University. He is working on a tool that can account for individual genetic variations when researching treatments and diseases. His work involves tackling how gene-expression-predictive models use genomic language models (gLMs). Liu’s work was recently published in npj Artificial Intelligence.
Most current gLMs rely on the “reference genome,” a standardized blueprint of human DNA assembled from multiple individuals. A different approach was needed to provide a better tool for individualized gene expression predictions.
“We pre-trained a powerful genomic language model (UKBioBERT) based on human variants from biobanks, and demonstrated that the embeddings from our model can enhance different expert models in performing gene expression prediction across individuals or genes,” said Liu. This language model was trained using real genetic variants from approximately 300,000 individuals in the UK Biobank, creating rich, function-aware representations of genomic sequences.
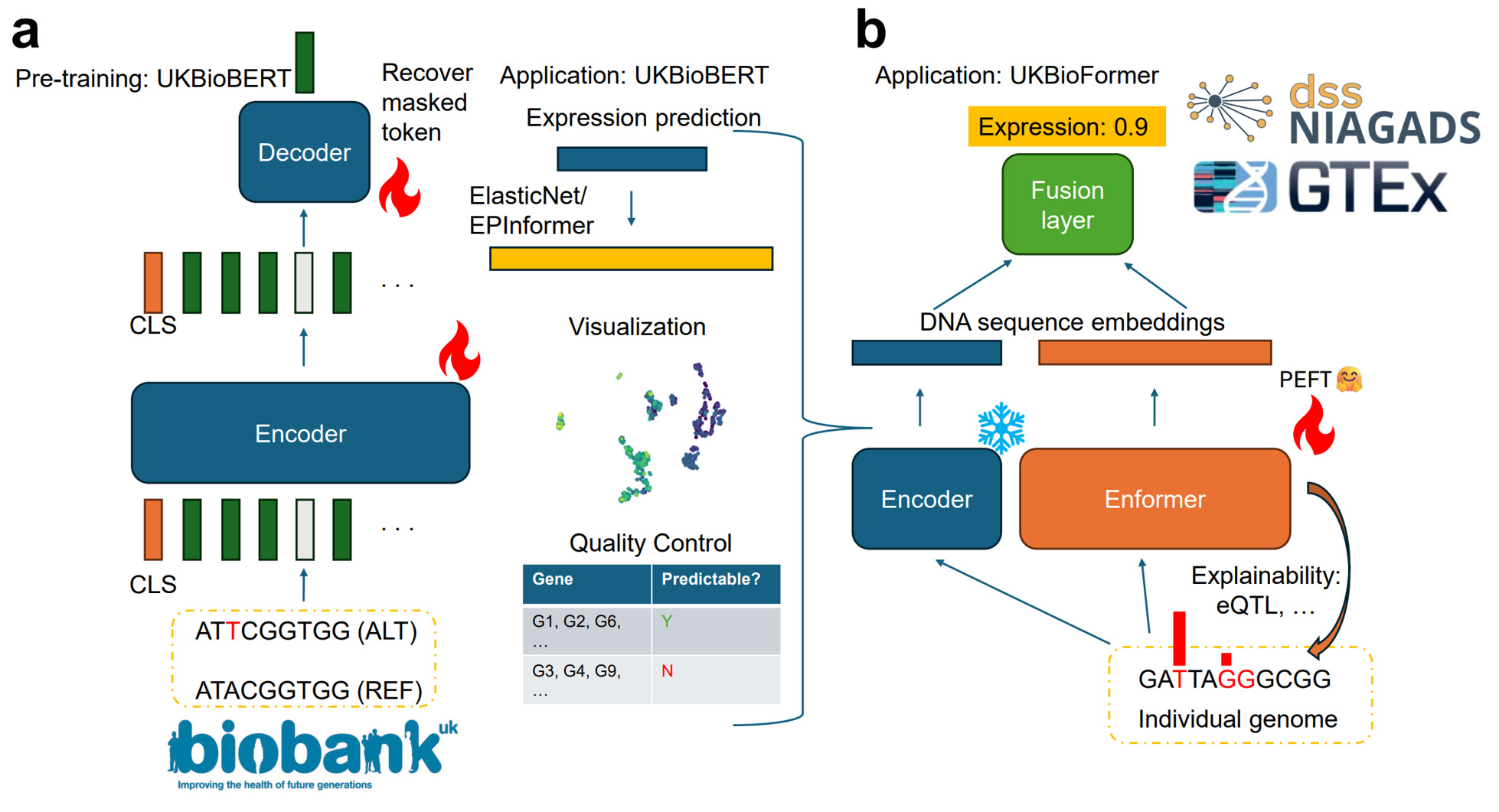
Building on this, the researchers created UKBioFormer and UKBioZoi by combining UKBioBERT with state-of-the-art architectures to improve the science of treatment and discovery. With these new tools, doctors would be better able to understand how your individual genes might affect things like disease risk or drug response. Conditions like cancer, diabetes, Alzheimer’s and autoimmune disorders are driven by subtle changes in gene expression rather than mutations in a single gene; these new tools help pinpoint those subtle regulatory effects. And, due to the broad dataset, results from these tools will be more applicable to a wider population with different ancestries than results based on a more limited reference genome, ensuring that treatments – from heart medication to weight-loss drugs – are tailored to the person, not the average.
NCSA resources provided valuable computation nodes for us to train a large model and conduct experiments efficiently. Previously, we needed to spend more than one month to train a model with 300 genes, but now we only need 10 days.
Yale University
Mapping the Biological Symphony

Xinyi Lisa Chen is a third-year Ph.D. student who also works in Professor Zhao’s lab. Chen is researching how genetic expression interacts with other parts of tissues. While Liu focuses on the unique ‘letters’ of an individual’s genetic code, Chen looks at how those instructions are carried out in physical space.
“Imagine watching the brain of a newborn mouse develop into adulthood,” said Chen, “cells gradually organizing into precise patterns, each performing distinct roles over time. To understand this biological symphony and discover how disruptions might lead to diseases like Alzheimer’s or Parkinson’s, scientists must piece together a puzzle involving not just what genes are active, but also where in the tissue they’re active, when they’re switched on, and how they interact with other biological processes.”
Scientists have recently been able to get unprecedented amounts of detailed information about cells during scans – sometimes they’re even able to isolate tiny groups of cells no bigger than two to three together to study them. “Specifically, scientists can measure both gene activity (RNA, or transcriptomics) and gene regulation (chromatin accessibility, or epigenomics via ATAC-seq) within these spots, while also precisely pinpointing their locations in specific regions,” Chen explained.
However, these snapshots of gene activity within cells had limitations. “Until now, scientists lacked a method to combine all these layers – spatial location, timing and multiple types of genetic data – into a single clear picture,” said Chen. To solve this, she created a tool called STORM (Spatial Temporal multi-Omics Representation Model). STORM uses graph neural networks to integrate these complex layers of information into one cohesive, biologically interpretable view.
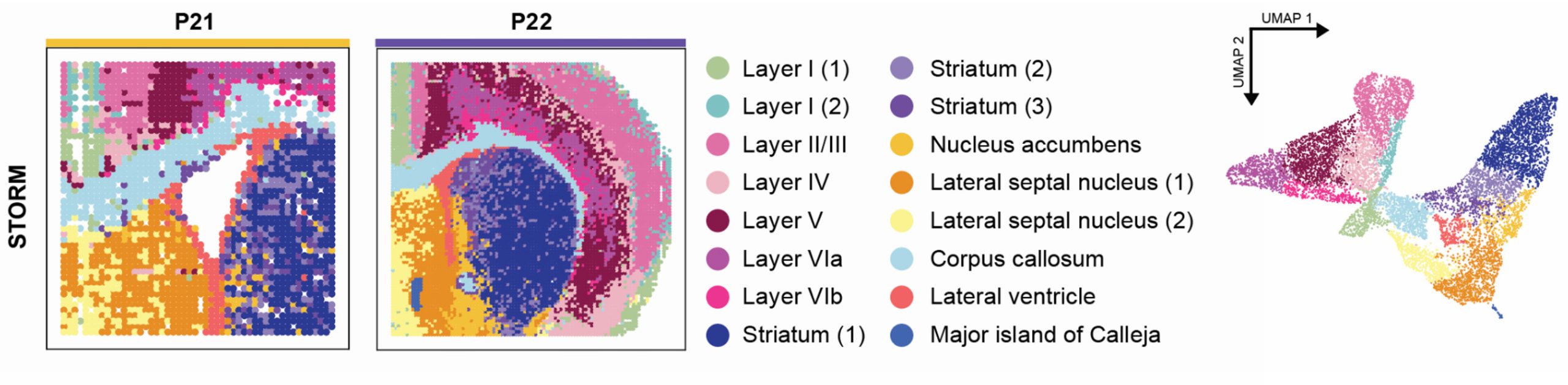
The hope is that this tool can be a valuable aid in personalized medicine. “While our current research involves mouse models, applying STORM to human atlas data can illuminate crucial developmental trajectories in tissues, highlighting pivotal moments when developmental processes may deviate from the norm,” Chen said. “Such insights could enable clinicians to administer targeted screenings or early interventions, significantly improving health outcomes. In elderly populations, STORM can help map the progression of neurodegenerative diseases like Alzheimer’s and Parkinson’s, potentially identifying critical windows for preventative strategies or therapeutic interventions, ultimately improving patient quality of life.”
The computational demands of STORM are substantial, given its application to vast datasets that integrate spatial information across multiple molecular layers and numerous developmental timepoints. High-performance computing resources provided by the NCSA were indispensable, particularly their advanced GPUs offering 96 GB of GPU memory.
Yale University
Liu and Chen were able to get time on NCSA’s Delta supercomputer through an allocation from the U.S. National Science Foundation ACCESS program. ACCESS helped connect these researchers to the computing power needed to turn complex AI theories into real biological discoveries. “Leveraging NCSA’s powerful H100 GPU, we successfully processed extensive datasets encompassing five timepoints and two modalities within just over 24 hours – a task previously infeasible even with other advanced GPUs like the A100,” said Chen. “This tremendous computational acceleration has allowed us to conduct research at a pace previously unattainable, rapidly advancing our understanding of complex biological processes.”
ABOUT DELTA AND DELTAAI
NCSA’s Delta and DeltaAI are part of the national cyberinfrastructure ecosystem through the U.S. National Science FoundationACCESS program. Delta (OAC 2005572) is a powerful computing and data-analysis resource combining next-generation processor architectures and NVIDIA graphics processors with forward-looking user interfaces and file systems. The Delta project partners with the Science Gateways Community Institute to empower broad communities of researchers to easily access Delta and with the University of Illinois Division of Disability Resources & Educational Services and the School of Information Sciences to explore and reduce barriers to access. DeltaAI (OAC 2320345) maximizes the output of artificial intelligence and machine learning (AI/ML) research. Tripling NCSA’s AI-focused computing capacity and greatly expanding the capacity available within ACCESS, DeltaAI enables researchers to address the world’s most challenging problems by accelerating complex AI/ML and high-performance computing applications running terabytes of data. Additional funding for DeltaAI comes from the State of Illinois.